
본 포스트는 LAIDD의 김동섭 교수님의 화학정보학 개론 수업을 기초로 작성되었습니다.
신약 개발 과정, 특히 Drug Discovery에서의 생명 & 화학정보학의 활용에 대해 알아본다.
신약 개발 과정은 다음과 같은 과정으로 나눌 수 있다.
- Drug Discovery
- Preclinical
- Clinical Trials(1,2,3)
- FDA Review
- LG-Scale MFG
Drug Discovery 단계에서 5000~10000가지의 후보물질이 Preclinical에서는 250가지, Clinical에서는 5가지, 마지막에는 한 가지로 결정되는 일련의 과정을 겪게 되고, 이는 10년이 넘는 시간이 소요된다.
하지만 다음 논문에서 주장하는 바와 같이,

시간이 갈수록 과거에 비해 같은 비용으로 찾을 수 있는 신약의 수가 감소하고 있다.
이는 R&D 효율이 과거에 비해 감소하고 있으며, 반도체 산업에서 흔히 말하는 Moore’s Law와
반대현상이라는 의미로 Eroom’s Law로 불린다.
이와 같은 현상의 원인으로 다음과 같은 사항들이 제기되었다.
- 기존의 약보다 더 나은 약효가 요구됨
- 과거 보다 강해진 식약처의 제한
- 기초 과학 연구의 부족
- 관심 질병이 제한됨
- 임상 과정의 문제
하지만 딥러닝과 같은 AI를의 활용과 생물&화학정보학 기법들을 통한 Drug Discovery가
신약 개발 과정을 단축시키고 개발 확률을 증가시키는 사례와 연구들이 등장하고 있다.
특히 개발 과정 중 첫번째 단계인 Drug Discovery 단계에서 그 활용이 기대된다.
해당 단계를 세분화하여 설명하고 각 단계에서 어떤 method가 활용 되는지 알아본다.
1. Tragent Identification
타겟 선정 단계로, 특정 질병과 연관되어 drug의 타겟이 될 signle molecule(주로 단백질)을 선정한다.
이때 “druggable” target을 찾는 것이 목표라는 표현이 자주 등장하게 된다.
“druggable”이란 drug 분자와 상호작용하고 영향을 받을 것으로 기대된다는 뜻이다.
computational method로 bioinformatics의 omics data 등을 이용하거나,
cheminformatics의 Reverse docking, Reverse QSAR가 활용된다.
Reverse docking의 dockingd은 원래 compound library에서 target과
binding을 잘하는 compound를 찾는 방식이라면(target → compound),
reverse docking은 역으로 여러 개의 target 중 천연물 혹은 phenotype 스크리닝으로 발견된 화합물과 binding이 좋은 target을 찾는 것이다.(compound → target)
2. Traget Validation
확인된 target이 질병에서 어떤 역할을 수행하는지 시험한다. 이 과정은 drug이 가질 의학적 효능과 직결된다.
Genetic knockdown, knockout, in vitro cell-based 메커니즘 연구 혹은 동물 실험이 진행된다.
3. Target to Hit
해당 과정부터 선도물질 발견을 위한 단계로 볼 수 있다.
선도 물질(Lead Compound)란 target에 영향을 주어 질병 치료를 기대할 수 있는 물질들을 뜻한다.
이 단계에서는 선도 물질 이전에 target과 binding 하는 물질들을 추리는 단계라고 할 수 있다.
High thrughput screening(HTS)은 target과의 반응이 예상되는 많은 물질들을 직접 반응시켜 확인하는 방법이다. 해당 방법은 전통적으로 사용되어 왔으나 시간과 비용이 많이 요구되는 작업이다.
HTS 대신 Computational method를 통해 더 효율적으로 Hit을 간추리는 것이 가능하다.
4. Hit to Lead
이전에 찾은 Hit들을 다시 Lead Compound로 간추리는 단계로, Lead generation으로 불리기도 한다.
선도물질을 찾기 위한 computational method로는 compound library design, virtual screening, docking, pharmacophore modeling, QSAR, De novo design, Deep learning 등이 사용된다.
Virtual Screening은 structure-based와 ligand-based로 나눌 수 있다.
structure-based의 경우, compound의 구조 정보만을 필요로 하며, 구조를 기반으로 target과의 binding을 예측하거나, docking 해보는 것을 말한다. 구조 정보만을 필요로 하기 때문에 해당 target과 binding하는 다른 물질의 정보가 필요하지 않아 새로운 target이 발견되었을 때, 사용할 수 있다는 장점이 있다. 단점으로는 뒤의 ligand-based 보다 정확도가 떨어진다.
ligand-based의 경우, 해당 target과 binding을 잘한다고 알려진 ligand들의 functional group에 대한 정보, 유사성 등을 비교하고 기계학습하는 방식으로 스크리닝을 진행하는 것을 뜻한다. 장점은 이미 binding을 잘하는 것으로 알려진 ligand들을 이용하기 때문에 정확도가 높다는 것이지만, 이미 알려진 ligand 들이 없을 경우 활용이 불가능하다는 단점이 있다.
5. Lead Optimization
Lead Compound의 구조를 변경하여 특성을 최적화 시키는 단계이다. 구조를 변화시킴으로써,
specificity를 증가시키거나 side effect를 줄이는 것을 목표로 한다.
computational method로는 QSAR, 3D-QSAR, structure-based optimization등을 활용하여 구조 변화 후 반응 변화를 예측한다.
6. Preclinical Testing
최적화된 Lead Compound를 실험, 동물 시험을 통해 임상을 적용할 수 있을지 확인하는 단계이다.
이때 safety test를 통해 약물의 ADME/T를 측정, 예측하게 된다.
여기서 ADME/T는 Absorption, Distribution, Metabolism, Excretion, Toxicity의 약자로,
- 혈류를 통한 absorption
- 체내 의도한 위치로의 distribution
- 효과적, 효율적인 metabolism
- 성공적은 체외 배출(excretion)
- 약물의 비독성의 증명(non-toxic)
위와 같은 사항들을 검사하는 것이 목적이며, 현재 동물, 세포실험 외에도 computational model을 이용한다.
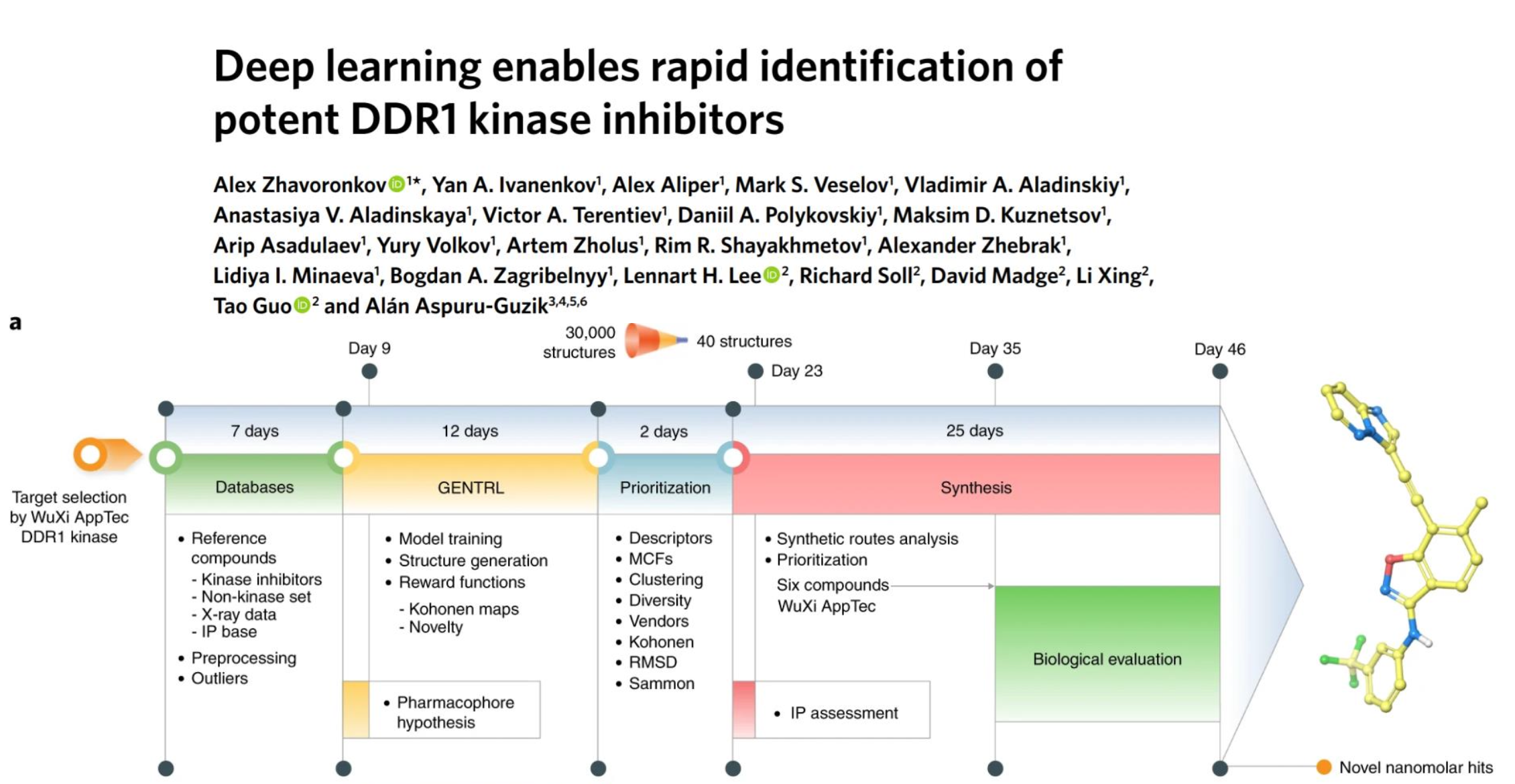
위 논문의 결과를 보면,
target selection 이후, promising hits를 찾는 과정이 Deep learning을 사용하여 단 46일만이 소요되었다.
실험적인 방법을 통해서는 5년 정도의 시간이 걸리던 것에 비해 굉장한 시간이 단축된다는 것을 알 수 있다.
COVID-19의 분석과 치료제 개발에 있어서도 빠른 대응을 위해 virtual screening이 적극적으로 활용되었다.
팬데믹 같은 긴급한 상황 속 빠른 시간 내 drug discovery가 가능하다는 것은 굉장히 중요한 성과이다.
추가적으로, R&D에 소요되는 비용과 시간을 낮추어 신약 개발의 빛을 받지 못하던 희귀병들의 연구와 개발이
활발히 진행되기를 기대한다.